Composto de coordenação
Os compostos de coordenação, são moléculas constituídas por um ou vários ácidos de Lewis ligados a uma ou várias bases de Lewis.[1]
Os ácidos de Lewis podem ser metais de transição e, neste caso, os compostos de coordenação também são chamados de complexos metálicos.
Exemplo:
- Cu2++4NH3⇌[Cu(NH3)4]2+{displaystyle Cu^{2+}+4NH_{3}rightleftharpoons [Cu(NH_{3})_{4}]^{2+}}
![Cu^{2+} + 4NH_3 rightleftharpoons [Cu(NH_3)_4]^{2+}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5f7536c21efe4e414677bfb10cd786f0ef926670)
No caso dos complexos metálicos, estes são compostos neutros resultantes da agregação de um complexo com um ânion. Um exemplo é o cloreto de hexaquocobre. As bases são chamadas de ligantes. Os ligantes são espécies ricas em elétrons, e os metais que formam complexos são íons com orbitais disponíveis para acomodar estes elétrons. A formação de complexos é comum com metais de transição d{displaystyle d}

Índice
1 Complexos metálicos
2 Termo coordenação
3 Teoria de Werner
4 Números atômicos efetivos
5 Tipos de ligantes
6 Desdobramento dos orbitais em campo Oh, Td e D4h
7 Energia de estabilização do campo cristalino
7.1 Campo fraco e campo forte
8 Teoria do campo cristalino
8.1 Magnetismo dos complexos
8.2 Cores dos complexos
9 Fatores que influenciam o desdobramento do campo cristalino
10 Estereoquímica dos Compostos de Coordenação de número 4
10.1 Geometria quadrado-planar
10.2 Geometria tetraédrica
10.3 Estereoquímica dos compostos de coordenação de número 5
11 Estereoquímica dos Compostos de Coordenação de número 6
12 Isomeria Estrutural
12.1 Isomeria de ligação
12.2 Isomeria de ionização
12.3 Isomeria de hidratação ou solvatação
12.4 Isomeria de coordenação
13 Isomeria geométrica e óptica em compostos de coordenação
13.1 Isomeria em complexos de número de coordenação quatro
13.2 Isomeria em complexos de número de coordenação cinco
13.3 Isomeria em complexos de número de coordenação seis
14 Reações em compostos de coordenação
14.1 Mecanismos de reações de substituição
14.2 Efeito trans
14.3 Estabilidade cinética e termodinâmica
14.4 Substituição em compostos octaédricos
14.5 Outros mecanismos de reação em compostos de coordenação – reações redox
15 Técnicas de caracterização de compostos de coordenação
15.1 Análise elementar (CHN) ou microanálise
15.2 Difratometria ou difração de raios X de pó (DRX)
15.3 Difratometria de raios X em monocristal
15.4 Espectroscopia eletrônica na região do UV/visível
15.5 Espectroscopia Vibracional na Região do Infravermelho e Espectroscopia Raman
15.6 Voltametria ou voltamperometria cíclica
16 Referências
17 Aplicações de compostos de coordenação
17.1 Ocorrência natural
17.1.1 Transporte de oxigênio
17.1.2 Fotossíntese
17.1.3 Vitamina B12
17.2 Extração de metais menos comuns
17.3 Pigmentos e corantes
17.4 Catálise
17.4.1 Ziegler
17.4.2 Wacker
17.4.3 Oxo
17.4.4 Ativação de pequenas moléculas
17.5 Química medicinal
18 Ver também
19 Bibliografia
20 Referências
Complexos metálicos |
Um complexo é um composto químico formado pela adição de uma substância simples, normalmente um íon metálico que funciona como um receptor de elétrons pi com uma ou várias moléculas de outra substância, chamada de ligante bases de Lewis.
Os casos mais comuns de complexos ocorrem com os compostos iônicos metálicos, que podem ter seus íons metálicos complexados tanto nos cristais quanto em solução.
Um exemplo típico é o sulfato de cobre, usado como anti-fungo em plantações. Seus cristais são azuis e os íons de cobre estão formando um complexo com a água no cristal. Uma vez aquecido a seco, perde água de cristalização e se torna branco.
Os indicadores de umidade que mudam de cor de azul a rosa contêm um sal de cobalto. O complexo de cobalto perde água de acordo com humidade do ar, mudando de cor.
Vários podem ser os ligantes: água, EDTA, monóxido de carbono, íons negativos (cloreto, iodeto, cianeto).
Estes ligandos podem ser classificados de acordo com o número de ligações que estabelecem ao átomo metálico central. Assim temos:
- 1 ligação: Ligando monodentado
- 2 ligações: Ligando bidentado
- 3 ou mais ligações: Ligando polidentado
Os complexos tem enorme importância na química e bioquímica, sendo praticamente todas as enzimas complexos de íons metálicos. Hemoglobina, clorofila e vitamina B, entre outros, são complexos respectivamente de ferro e magnésio e cobalto.
Termo coordenação |
No contexto da química de coordenação, o termo complexo significa um átomo metálico ou íon central rodeado por um conjunto de ligantes.[2] Um ligante é um íon ou molécula que pode ter existência independente. Exemplo de um complexo é o [Co(NH3)6)]3+{displaystyle [Co(NH_{3})_{6})]^{3+}}![[Co(NH_3)_6)]^{3+}](https://wikimedia.org/api/rest_v1/media/math/render/svg/cb476a016bacaf6c645f7ecf8f3efc414e4d6e2b)


Teoria de Werner |
As principais características das estruturas geométricas dos complexos metálicos foram identificadas por Alfred Werner (1866-1919), profundo conhecedor da estereoquímica orgânica. A teoria da coordenação de Werner, de 1893, foi a primeira tentativa de explicar a ligação existente em complexos de coordenação. Esta teoria foi proposta antes da descoberta do elétron por J.J. Thompson em 1896, e antes da formulação da teoria eletrônica de valência. Essa teoria, e os 20 anos de trabalhosas pesquisas associados a ela, deu a Werner o prêmio Nobel de Química de 1913.
Werner não tinha à sua disposição nenhuma das modernas técnicas instrumentais, e todos os seus estudos foram feitos mediante a interpretação de simples reações químicas. Werner foi capaz de explicar as principais características das estruturas geométricas dos complexos metálicos, e concluiu que nos complexos o metal apresenta dois tipos de valência. Valência primária corresponderia ao número de carga do íon complexo, hoje chamado estado de oxidação. Valência secundária corresponderia ao número de ligantes coordenados ao metal, hoje chamado de número de coordenação.
Werner combinou a interpretação de isomerismo óptico e geométrico com padrões de reações e com dados de condutividade elétrica num trabalho que ainda permanece como modelo de como usar, de maneira efetiva e criativa, evidências físicas e químicas. As cores marcantes de muitos compostos de coordenação de metais d{displaystyle d}
Com a teoria de Werner foi possível verificar que um número grande de moléculas e íons pode se comportar como ligantes, e um grande número de íons forma complexos.
A teoria da coordenação de Werner, de 1893, foi a primeira tentativa de explicar a ligação existente nos complexos de coordenação. Ele concluiu que esses compostos apresentam dois tipos de valência:
Valência primária: é o número de cargas no íon complexo. Por exemplo, no composto CoCl2{displaystyle CoCl_{2}}, temos Co2+{displaystyle Co^{2+}}
e mais dois átomos de Cl−{displaystyle Cl^{-}}
, portanto, temos 2 valências primárias.
Valência secundária: Se refere ao número de átomos ligantes coordenados ao metal central, ou seja, é o Número de Coordenação do composto. Cada ligante doa um par de elétrons ao íon metálico, formando uma ligação coordenada.
Números atômicos efetivos |
Sidwick sugeriu que os pares de elétrons dos ligantes são adicionados ao metal até que este esteja rodeado por números de elétrons equivalentes ao do gás nobre mais próximo. Essa regra prevê o número de ligantes em muitos complexos, mas há exceções. Não deve-se tomar como regra absoluta para a formação de um composto coordenado.
Tipos de ligantes |
Os ligantes podem ser classificados conforme o número de átomos ligados ao íon metálico. Ligantes que se coordenam através de um átomo são chamados de monodentados. Ligantes que possuem mais do que um ponto de ligação são chamados como polidentados. Os ligantes que se coordenam através de dois átomos são chamados de bidentados, aqueles com três, como tridentados e assim por diante.
Um exemplo é o ligante bidentado etilenodiamino (em, NH2CH2CH2NH2{displaystyle NH_{2}CH_{2}CH_{2}NH_{2}}

O ligante hexadentado, o ácido etilenodiaminotetraacético, na forma do seu ânion [(EDTA), (−O2CCH2)2NCH2CH2N(CH2CO2−)2]{displaystyle ({}^{-}O_{2}CCH_{2})_{2}NCH_{2}CH_{2}N(CH_{2}CO_{2}{}^{-})_{2}]}![({}^-O_2CCH_2)_2NCH_2CH_2N(CH_2CO_2 {}^-)_2]](https://wikimedia.org/api/rest_v1/media/math/render/svg/37cff6f2133f90e847f91a5aa6174a5ab7e326d6)



Ligantes ambidentados são aqueles que têm, potencialmente, mais de um átomo doador diferente. Um exemplo é o íon tiocianato (NCS−{displaystyle NCS^{-}}

Os ligantes podem ser classificados também de acordo com a sua carga, ligantes negativos (aniônicos), ligantes neutros (moléculas) e ligantes positivos (catiônicos). Os ligantes positivos são muito raros.
Segue alguns exemplos:
São numerosos os íons negativos que desempenham o papel de ligantes em compostos de coordenação. Eles podem ser classificados em dois tipos:
Monodentados: Ocupam um sítio de coordenação, onde os ligantes podem se ligar. Exemplos são os íons fluoreto, cloreto, cianeto e ligantes neutros, como a amônia e a água.
Polidentados: Ocupam mais de um sítio de coordenação. São chamados de Quelatos.
Quelatos: Quando o ligante ocupa mais de um sítio, formando assim, uma estrutura cíclica. Estes compostos são mais estáveis que os monodentados. Quanto maior o número de anéis formados, maior será a estabilidade do composto. Exemplos: Etilenodiamina (en), (EDTA).
Desdobramento dos orbitais em campo Oh, Td e D4h |
A teoria do campo cristalino considera que a interação entre os ligantes e o íon metálico em complexos é de natureza puramente eletrostática e prevê que os níveis de energia dos orbitais d{displaystyle d}
Considerando-se um íon metálico M+{displaystyle M^{+}}

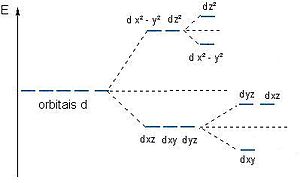
Alguns orbitais d{displaystyle d}

Os orbitais dz2{displaystyle dz^{2}}






Com raciocínio análogo, analisando uma estrutura tetraédrica, conforme figura 3 e 4, observa-se que os orbitais dxy,dzx,dyz{displaystyle dxy,dzx,dyz}

Tendo em vista as observações acima temos o diagrama do nível de energia abaixo para sistemas tetraédricos:
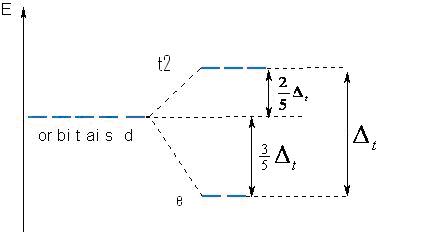
Onde e refere-se aos orbitais dz2{displaystyle dz^{2}}

Quando as distâncias cátion-ânion são iguais e o cátion e o anion são os mesmos nos casos octaédrico e tetraédrico tem-se que Δt=49Δ0{displaystyle Delta _{t}={tfrac {4}{9}}Delta _{0}}
Alguns complexos costumam desviar-se da geometria octaédrica apresentando distorções tetragonais, como por exemplo, complexos hexacoordenados de cobre(II). Essa distorção é conhecida como distorção Jahn-Teller e corresponde a uma extensão ao longo do eixo z{displaystyle z}



Alongamento dos ligantes ao longo do eixo:

Compressão dos ligantes ao longo do eixo z{displaystyle z}
Quanto aos complexos de geometria quadrada planar, típicos D4h, conforme apresentado na figura 8, observa-se que deriva de uma estrutura octaédrica considerando os ligantes ao longo do eixo z{displaystyle z}

Energia de estabilização do campo cristalino |
O principal efeito dos ligantes, segundo a Teoria do campo Cristalino (TCC), é o desdobramento dos níveis de energia correspondentes aos orbitais d{displaystyle d}
A diferença de energia entre os dois conjuntos de orbitais denomina-se desdobramento do campo cristalino ou desdobramento do campo ligante. Este desdobramento é representado pelo parâmetro do desdobramento do campo cristalino Δ{displaystyle Delta }





- EECL=(0,4x−0,6y)Δo{displaystyle EECL=(0,4x-0,6y)Delta _{o}}

O termo energia de estabilização do campo cristalino (EECC) é muito usado no lugar de EECL, mas esse termo é mais apropriado para íons nos cristais.
A tabela abaixo (tabela 1) apresenta os valores de EECL para diferentes configurações. A EECL é geralmente apenas uma pequena fração da interação total entre o átomo metálico e os ligantes, a qual aumenta da esquerda para a direita ao longo de um período devido ao decréscimo no raio dos íons M2+{displaystyle M^{2+}}
Tabela 1 Energia de Estabilização do campo Ligante
Campo fraco e campo forte |
Para um íon d4{displaystyle d^{4}}








Teoria do campo cristalino |
A EECC é igual a zero para íons com configuração d0 e d10{displaystyle d^{0}~e~d^{10}}

Complexos octaédricos são geralmente mais estáveis e mais comuns que os complexos tetraédricos, já que a EECC de um complexo octaédrico é maior que a EECC de um complexo tetraédrico, quando se considera o mesmo íon metálico e os mesmos ligantes.
O estudo quantitativo da estabilidade dos complexos e quelatos pode ser feito através do uso da "constante de estabilidade" ou da "constante de formação" desses compostos. A estabilidade dos complexos é determinada pela energia de ligação metal-ligante (M−L{displaystyle M-L}
- My++xL→[MLx]y+{displaystyle M^{y+}+xLrightarrow [MLx]^{y+}}
![M^{y+} + xL rightarrow [MLx]^{y+}](https://wikimedia.org/api/rest_v1/media/math/render/svg/bab1fb2115a9e1c76aca95f57e769e8db380d47d)
onde, My+{displaystyle M^{y+}}

![[MLx]^{y+}](https://wikimedia.org/api/rest_v1/media/math/render/svg/95fc8150ff1363cce0573ad8cda72a511b90c238)
Magnetismo dos complexos |
Uma espécie paramagnética possui elétrons desemparelhados e sofre atração por um campo magnético. Já uma substância é considerada diamagnética quando não possui elétrons desemparelhados e sofre repulsão de um campo magnético. Grande parte dos complexos de metal d{displaystyle d}
Os ligantes de campo forte produzem uma grande diferença de energia entre os orbitais t2g e eg{displaystyle t_{2g}~e~e_{g}}

Ligantes de campo fraco provocam pequena diferença de energia entre t2g e eg{displaystyle t_{2g}~e~e_{g}}
Nas configurações eletrônicas d1,d2,d3,d8 e d9{displaystyle d^{1},d^{2},d^{3},d^{8}~e~d^{9}}
Cores dos complexos |
A luz branca é constituída pela soma de todos os comprimentos de onda do espectro visível, que vai de 380 à 720 nm aproximadamente. Quando a energia correspondente a algum desses comprimentos de onda é absorvida por um complexo para proporcionar transições eletrônicas, vemos a cor complementar àquela responsável pelo comprimento de onda absorvido. Assim é possível determinar experimentalmente o valor de ∆o para a maioria dos complexos a partir do seu espectro de absorção. Por exemplo: o íon hexaaquotitânio(III), [Ti(H2O)6]3+{displaystyle [Ti(H_{2}O)_{6}]^{3+}}![[Ti(H_2O)_6]^{3+}](https://wikimedia.org/api/rest_v1/media/math/render/svg/51d6b247e8ed7cf9e7555f3e892a1551408e0ce2)


Fatores que influenciam o desdobramento do campo cristalino |
A separação dos dois conjuntos de orbitais de energias diferentes nos complexos é chamada de parâmetro do desdobramento do campo ligante (10Dq{displaystyle 10Dq}
São eles:
- a) Simetria de campo
Quanto maior o número de ligantes, mais forte é o campo, pois o valor de 10 Dq depende do número de ligantes e de seu arranjo em torno do átomo metálico. Assim, um complexo octaédrico terá sempre um campo mais forte do que um tetraédrico formado pelas mesmas espécies de ligantes e metais. Os compostos tetraédricos são sempre de campo fraco.
- b) Número de oxidação do metal
Quanto maior for o número de oxidação do metal maior o valor de 10 Dq. Isso acontece porque uma carga positiva elevada no íon metálico fará com que ele atraia fortemente os ligantes aniônicos ou polares, aumentando a interação eletrostática entre eles e os elétrons nos orbitais d. Essa variação também reflete o tamanho menor dos íons de maior carga e, conseqüentemente as menores distâncias metal-ligante resultando em energias de interação mais fortes. Para os metais de transição da primeira série, os valores de ∆o para um metal com número de oxidação +3 são, aproximadamente, 50% maiores do que para um metal com número de oxidação +2.
- c) Identidade do metal
O valor do desdobramento do campo aumenta significativamente à medida que se desce num mesmo grupo da tabela periódica. Isso ocorre devido ao tamanho maior dos átomos dos orbitais 4d e 5d em relação aos orbitais 3d, o que aumenta a interação com os ligantes. Por isso, a maioria dos complexos do 2º e 3º períodos é de campo forte. A força do campo ligante tem sua ordem crescente de energia (aproximadamente) apresentada abaixo:
- Mn+2<Ni+2<Co+2<Fe+2<Co+3<Mo+3<Rh+3<Ru+3<Pd+4<Ir+3<Pt+4{displaystyle Mn^{+2}<Ni^{+2}<Co^{+2}<Fe^{+2}<Co^{+3}<Mo^{+3}<Rh^{+3}<Ru^{+3}<Pd^{+4}<Ir^{+3}<Pt^{+4}}
- Mn+2<Ni+2<Co+2<Fe+2<Co+3<Mo+3<Rh+3<Ru+3<Pd+4<Ir+3<Pt+4{displaystyle Mn^{+2}<Ni^{+2}<Co^{+2}<Fe^{+2}<Co^{+3}<Mo^{+3}<Rh^{+3}<Ru^{+3}<Pd^{+4}<Ir^{+3}<Pt^{+4}}

- d) Natureza do ligante
O parâmetro de desdobramento do campo ligante varia de acordo com a natureza do ligante. Verificou-se que determinados ligantes provocam um maior desdobramento de campo do que outros, ou seja, aumenta a energia da transição e a luz absorvida terá um comprimento de onda menor, resultando em diferentes cores para os respectivos complexos. Dados experimentais evidenciaram que independente da identidade do íon metálico a mesma ordem é seguida. Ryutaro Tsuchida propôs organizar os ligantes em ordem crescente de energia das transições a qual chamou de série espectroquímica:
- I−<Br−<S−2<SCN−<Cl−<NO2−<N3−<F−<OH−<C2O4−2<H2bO<{displaystyle I^{-}<Br^{-}<S^{-2}<SCN^{-}<Cl^{-}<NO_{2}{}^{-}<N_{3}{}^{-}<F^{-}<OH^{-}<C_{2}O_{4}{}^{-2}<H_{2}bO<}
- <NCS−<CH3CN<py<NH3<en<bipy<phen<NO−2<PPh3<CN−<CO{displaystyle <NCS^{-}<CH_{3}CN<py<NH_{3}<en<bipy<phen<NO^{-2}<PPh_{3}<CN^{-}<CO}
- I−<Br−<S−2<SCN−<Cl−<NO2−<N3−<F−<OH−<C2O4−2<H2bO<{displaystyle I^{-}<Br^{-}<S^{-2}<SCN^{-}<Cl^{-}<NO_{2}{}^{-}<N_{3}{}^{-}<F^{-}<OH^{-}<C_{2}O_{4}{}^{-2}<H_{2}bO<}


Os átomos sublinhados são os doadores nos complexos ambidentados. Como se pode observar, o desdobramento provocado, por exemplo, pelo ligante CN−{displaystyle CN^{-}}
Estereoquímica dos Compostos de Coordenação de número 4 |
Estereoquímica do complexo é a relação espacial entre um íon metálico central e seus ligantes. A estereoquímica pode ser agrupada de acordo com o número de coordenação (NC) da espécie central; no caso o número de coordenação 4 significa que possuímos um íon central ligado a mais quatro elementos, iguais ou diferentes entre si. Moléculas de íons poliatômicos que possuem a mesma fórmula molecular, mesmas ligações, mas diferentes estruturas e arranjos espaciais são chamados de estereoisômeros. A distribuição dos ligantes ao redor do íon central está coordenada de acordo com a repulsão mútua entre os ligantes e o impedimento estérico dos ligantes polidentados. O arranjo dos ligantes ao redor do íon central é influenciado por:
- Tamanho do ligante.
- Natureza do ânion.
- Procedimento de síntese utilizado para obtenção do complexo.
Existem duas geometrias comuns associadas com um número de coordenação igual a quatro. Essas geometrias são a quadrado-planar e a tetraédrica. Cada uma destas geometrias permite uma forma diferente de estereoisomerismo.
Geometria quadrado-planar |
Os complexos que apresentam essa geometria estão caracterizados pela configuração d8{displaystyle d^{8}}




[Pt(NH3)4]2+,[PtCl2(NH3)2],[Ni(CN)4]2−,[AgF4]−,[Cu(NH3)4]2+{displaystyle [Pt(NH_{3})_{4}]^{2+},[PtCl_{2}(NH_{3})_{2}],[Ni(CN)_{4}]^{2-},[AgF_{4}]^{-},[Cu(NH_{3})_{4}]^{2+}}.
Geometria quadrado-planar pode também ser forçada em um átomo central pela complicação com um ligante que contem um anel rígido de quatro átomos doadores.
Quando os ligantes estão aos pares diferindo apenas na posição do arranjo consideramos a isomeria cis e trans:
Geometria tetraédrica |
Complexos tetraédricos de simetria aproximadamente Td são favorecidos em números altos de coordenação quando o átomo central é pequeno e os ligantes são grandes (como Cl−,Br− e I−{displaystyle Cl^{-},Br^{-}~e~I^{-}}




Os complexos que apresentam essa geometria também como a geometria quadrado- planar; caracterizam-se por distribuições eletrônicas d8{displaystyle d^{8}}![[BF_4]^-, [MnO_4]^-, [ZnCl_4]^{2-}, [Zn(NH_3)_4]^{2+}](https://wikimedia.org/api/rest_v1/media/math/render/svg/68f9d6c54f123570394c7520c735371795bb1a4c)

Os enantiômeros puros ou em solução possuem a propriedade de girar o plano da luz polarizada. Essas substâncias que são capazes de girar o plano da luz polarizada são chamadas de opticamente ativas. A síntese de complexos tetraédricos do tipo MABDC é difícil e geralmente conduz à formação de uma mistura de dois enantiômeros. As espécies quirais nestes complexos existem quase sempre em um rápido equilíbrio de interconversão, na medida em que as ligações metal-ligante são rapidamente desfeitas e refeitas (os complexos são considerados lábeis).
Ocorre, portanto, que o enantiomerismo é raramente observado nos complexos tetraédricos simples.
Caso especial:

Estereoquímica dos compostos de coordenação de número 5 |
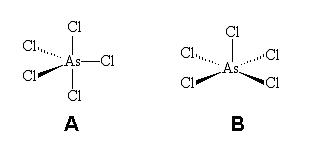
Complexos pentacoordenados, que são menos comuns do que os complexos tetra ou hexacoordenados no bloco d, são pirâmides de base quadrada ou bipirâmide trigonal.
Entretanto, distorções destas geometrias ideais são comuns. A forma bipiramidal trigonal, minimiza as repulsões ligante-ligante, mas restrições estéricas em ligantes polidentados podem favorecer uma estrutura piramidal quadrada. Por exemplo, a pentacoordenação piramidal quadrática é encontrada entre as porfirinas biologicamente importantes, onde o anel ligante obriga uma estrutura quadrada-planar e um quinto ligante preso acima do plano. A estrutura abaixo mostra o centro ativo de mioglobina, a proteína de transporte de oxigênio; a localização do átomo de ferro acima do plano do anel é importante para a sua função.[3]
Centro ativo de Mioglobina: Proteína de transporte de oxigênio.
Em alguns casos, a pentacoordenação é induzida por um ligante polidentado contendo um átomo doador que pode ligar-se em um sítio axial de uma bipirâmide trigonal, com seus átomos doadores remanescentes alcançando até as posições equatoriais como no exemplo abaixo.
As energias das várias geometrias de complexos pentacoordenados frequentemente diferem pouco uma da outra. A delicadeza deste balanço destaca-se pelo fato de que [Ni(CN)5]3−{displaystyle [Ni(CN)_{5}]^{3-}}![[Ni(CN)_5]^{3-}](https://wikimedia.org/api/rest_v1/media/math/render/svg/15c4000c7c12dfdd3c651e4f02f5700c7680844b)
Uma pseudo-rotação de Berry, onde (a) [Fe(CO)5]{displaystyle [Fe(CO)_{5}]}![[Fe(CO)_5]](https://wikimedia.org/api/rest_v1/media/math/render/svg/e66e4e3464d0090f054f6cf2d03db0fce74d85b8)
Estereoquímica dos Compostos de Coordenação de número 6 |
A grande maioria dos compostos hexacoordenados apresenta geometria octaédrica. O arranjo octaedrico é bastante simétrico, sendo o ponto de partida para entender outros arranjos deste grupo, que são distorçoes do octaedro, em função do fator de empacotamento, ou seja, uma compressão ou alongamento ao longo dos eixos de ordem dois, três ou quatro, como mostrado a seguir. Estas distorções podem ser tetragonais (mais simples), de forma que os ligantes sobre o eixo diferem dos outros quatro, para mais ou para menos, ou pode haver distorções rômbicas, nas quais um ligante trans está mais próximo do átomo central enquanto o outro está mais afastando. Estas distorções geram uma família imensa de isômeros geométricos que variam do octaedro ao prisma trigonal (D3h).[3]
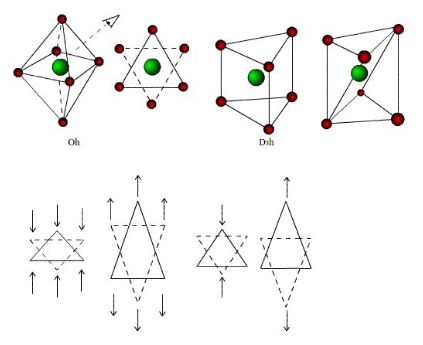
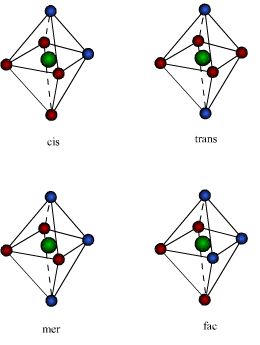
Os compostos octaedricos permitem uma variedade de estereoisômeros. A partir do fato que os seis vértices de um octaedro são equivalentes, apenas uma estrutura é possível para complexos dos tipos MA6 e MA5B. Já para o MA4B2 podem existir isômeros cis e trans. No isômero cis os dois ligantes B ocupam os vértices adjacentes do octaedro; no isômero trans estão em vértices opostos, como mostra a figura.Para os complexos do tipo MA3B3, também são possíveis dois isômeros, denominados: facial (fac) e meridional (mer):
Caso o número de ligantes aumente ou então se considerarmos ligantes polidentados, mais casos de isomerismos geométricos podem existir, mas estes devem ser relacionados com os isômeros cis, trans, mer, fac, utilizando uma numeração para explicar.
Os compostos octaedricos também podem apresentar geometria óptica, apresentando enantiômeros opticamente ativos, ou misturas racêmicas. Sendo todos os ligantes complexados ao átomo central diferentes, obviamente o composto apresenta geometria óptica. Há diversas possibilidades de isômeros ópticos no octaedro variando o número de ligantes diferentes, sendo ou não anéis quelantes. O mais comum são isômeros com anéis quelantes, devido a maior estabilidade e isômeros não quelantes são difíceis de sintetizar e são diversos os enantiômeros. Os enantiômeros podem ser mais bem observados analisando a vista fácil do tetraedro.
Isomeria Estrutural |
Uma das classes de isômeros é a isomeria estrutural, que compreende compostos com mesmos átomos e que não podem ser distinguidos apenas por sua formula molecular. Algumas das formas desta isomeria são a de ligação, de ionização, de hidratação e a de coordenação.
Isomeria de ligação |
A isomeria de ligação ocorre quando um ligante monodentado possui mais de um átomo com par de elétrons isolado que pode ligar-se ao íon do metal, mas devido ao seu tamanho ou forma, somente um átomo de cada vez pode ligar-se ao metal. Os ligantes mais comuns que apresentam este isomerismo são: SCN−{displaystyle SCN^{-}}


(a) o ligante, neste caso, é o NCS−{displaystyle NCS^{-}}
Isomeria de ionização |
Ocorre quando o contra-íon no sal complexo é um pré-ligante e pode deslocar um ligante, que se torna então o contra-íon , por exemplo:
- Os sais [Co(NH3)5SO4]Br{displaystyle [Co(NH_{3})_{5}SO_{4}]Br}![[Co(NH_3)_5SO_4]Br](https://wikimedia.org/api/rest_v1/media/math/render/svg/7ad41fcbaf8f9092d067090d8df056494e356a41)
![[Co(NH_3)_5Br]SO_4](https://wikimedia.org/api/rest_v1/media/math/render/svg/cc731b5f1980b8462ed2702482f345c30600744e)
Isomeria de hidratação ou solvatação |
É um tipo especial de isomeria, em que a água está envolvida e é de certa forma semelhante à isomeria de ionização. Diferem pela troca entre a molécula de H2O{displaystyle H_{2}O}
![[Cr(H_2O)_6]Cl_3](https://wikimedia.org/api/rest_v1/media/math/render/svg/8d562ab06e44fba12664a37d54e5f5a7f98522e1)
![[CrCl(H_2O)_5]Cl_2.H_2O](https://wikimedia.org/api/rest_v1/media/math/render/svg/0ae18b03bd9af64948f4b033f3f03f5642d7db2a)
![[CrCl_2(H_2O)_4]Cl.2H_2O](https://wikimedia.org/api/rest_v1/media/math/render/svg/a89b4f6694e0252361592d50b0a940c6fcb3601e)
Isomeria de coordenação |
Ocorre quando um ou mais ligantes são trocados entre os cátions e ânions complexos. Um exemplo de um par de isômeros de coordenação é:
Hexacianoferrato(III) de hexaamincromo(III), [Cr(NH3)6][Fe(CN)6]{displaystyle [Cr(NH_{3})_{6}][Fe(CN)_{6}]}![[Cr(NH_3)_6][Fe(CN)_6]](https://wikimedia.org/api/rest_v1/media/math/render/svg/b615a92803400fca764013d6b001d61874e29f77)
![[Fe(NH_3)_6][Cr(CN)_6]](https://wikimedia.org/api/rest_v1/media/math/render/svg/bb3742b1f962d828e4bab5e959ca44270eaf4952)
Um caso especial de isomeria de coordenação é aquela apresentada por uma série de compostos de mesma fórmula empírica, porém de diferentes massas moleculares. A este tipo de isomeria se dá o nome de "isomeria de polimerização", apesar dela não envolver polimerização de acordo com sua definição convencional. Como exemplo temos a série de sais nos quais tanto o cátion quanto o ânion contém Co3+{displaystyle Co^{3+}}
[Co(NH3)6][Co(NO2)6]{displaystyle [Co(NH_{3})_{6}][Co(NO_{2})_{6}]}e o [Co(NH3)4(NO2)2][Co(NH3)2(NO2)4]{displaystyle [Co(NH_{3})_{4}(NO_{2})_{2}][Co(NH_{3})_{2}(NO_{2})_{4}]}
![[Co(NH_3)_4(NO_2)_2][Co(NH_3)_2(NO_2)_4]](https://wikimedia.org/api/rest_v1/media/math/render/svg/6d09f1d934c28bc6a34cf94b280ec61d3edc2f93)
Isomeria geométrica e óptica em compostos de coordenação |
O critério formal de quiralidade é ausência de um eixo de rotação impróprio (Sn{displaystyle S_{n}}


Isomeria em complexos de número de coordenação quatro |
Complexos tetracordenados possuem duas estruturas geométricas principais: o tetraédro e o quadrado planar.
As espécies que apresentam a geometria quadrado-planar (Ex. [PtCl4]2−{displaystyle [PtCl_{4}]^{2-}}![[PtCl_4]^{2-}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d8284d10f58a188427ddf07ef201e4288b06b099)
![[Ni(CN)_4]^{2-}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8c3c5e3e92b26b26ecb4aa5d54bc400e068dc152)

As espécies que apresentam a geometria tetraédica (Ex.: [CoCl4]2−,[CrO4]2−{displaystyle [CoCl_{4}]^{2-},[CrO_{4}]^{2-}}![[CoCl_4]^{2-}, [CrO_4]^{2-}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8233c8712afcdaf6078bdb7b1b3d4cc22ce191c9)
Complexos com geometria tetraédrica podem apresentar isomeria óptica de forma semelhante aos compostos orgânicos. Logo, a forma mais simples de apresentar este comportamento é possuir os quatro ligantes diferentes entre si. Todavia, em alguns destes complexos a dificuldade de separação é tão grande que se considera que alguns não são opticamente ativos, pois interconvertem, formando mistura racêmica. Um exemplo de composto tetraédrico que possui isomeria óptica são os enantiômeros do bis(benzoilacetonato) de berílio.
Os compostos quadrados planares raramente mostram tal isomeria. O plano formado pelos quatro átomos ligantes e o íon metálico central geralmente é um plano especular que impede a possibilidade de assimetria óptica. Um exemplo de composto quadrado planar que possui isomeria óptica é o (mesobendiamina)(isobutilediamina)paládio(II)
Isomeria em complexos de número de coordenação cinco |
Espécies contendo este número de coordenação são mais raras do que aquelas com número de coordenação igual a quatro e seis. Existem duas possibilidades de geometrias moleculares, a bipirâmide trigonal (A{displaystyle A}

[Ni(CN)5]−{displaystyle [Ni(CN)_{5}]^{-}}e CoBrN(CH2CH2NMe2)3{displaystyle CoBrN(CH_{2}CH_{2}NMe_{2})_{3}}
.
Materiais compostos por moléculas que possuem o átomo central pentacoordenado apresentam as duas geometrias moleculares possíveis simultaneamente. Isto ocorre devido a um fenômeno chamado Pseudo-Rotação de Berry, representada na figura abaixo:

Em complexos com número de coordenação cinco é sabido que teoricamente possuem diversas geometrias possíveis para isomeria óptica, porém conhecem-se poucos exemplos. Um exemplo de isômero com esta estrutura é o cátion dicarbonilpentadienil-molibdenio (II):
Isomeria em complexos de número de coordenação seis |
A coordenação octaédrica é a mais comum e permite diversos tipos de estereoisomerismo. Supondo-se que os seis vértices de um octaedro são equivalentes:
- Apenas uma estrutura é possível para complexos dos tipos MA6 e MA5B{displaystyle MA_{6}~e~MA_{5}B}
, não possuindo isômeros geométricos.
- Para o MA4B2{displaystyle MA_{4}B_{2}}
, contudo, as estruturas cis e trans podem existir. No isômero cis os dois ligantes B{displaystyle B}

- Para os complexos do tipo MA3B3{displaystyle MA_{3}B_{3}}
, dois isômeros geométricos são possíveis e são denominados facial (fac) e isômero meridional (mer). As faces, que dão nome ao isômero geométrico fac, estão representadas em I e II na figura abaixo:


Na geometria octaédrica diferentes arranjos de ligantes podem dar origem a isômeros ópticos. Um exemplo simples é o [Mn(acac)3]{displaystyle [Mn(acac)_{3}]}![[Mn(acac)_3]](https://wikimedia.org/api/rest_v1/media/math/render/svg/5171b9b8f70e4ee2644c9f4b2094708eba046855)
![[MA_2B_2C_2]](https://wikimedia.org/api/rest_v1/media/math/render/svg/8e322c53409352d308231f22b37c4ec4f9433cb2)
Reações em compostos de coordenação |
Mecanismos de reações de substituição |
A Cinética_química está relacionada com a velocidade de reação. Compostos de coordenação que sofrem reação com tempo menor que 1 minuto são descritos como lábeis. Se a reação do complexo metálico demorar mais que 1 minuto, o composto é considerado inerte, essa é a regra de ouro de Taube. Assim, os termos lábil e inerte estão relacionados com a cinética.[4] A cinética é importante para compreender os mecanismos de reação e, no caso de complexos metálicos ou compostos de coordenação, é possível comparar a velocidade de substituição com a estrutura eletrônica dos íons metálicos, estudando a influência dessa estrutura na velocidade de substituição.[5] Reações e mecanismos de substituição de complexos inorgânicos foram muito estudados e uma revisão bastante interessante, em que os termos lábil e inerte foram utilizados pela primeira vez, foi feita por Taube em 1951.[6] Em química de coordenação, reações de substituição de ligantes são definidas como a reação geral a seguir:
M-L + X → M-X + L
O ligante L, que estava coordenado ao metal, é substituído pelo ligante X sem a mudança no estado de oxidação do metal. O ligante L é denominado ligante de saída, enquanto que o ligante X é denominado ligante de entrada. De acordo com o estudo das velocidades de reação (taxa de reação), é possível definir a etapa determinante da velocidade (etapa lenta) com relação à quebra ou formação de ligação. Dessa forma, os mecanismos de reações de substituição em complexos metálicos podem ser separados em associativo (A), dissociativo (D) e de intercâmbio (I).
Em reações químicas os reagentes se transformam em produtos passando por um estado de transição. Em alguns casos é possível que haja a formação de um intermediário nesse estado de transição. Quando esse intermediário formado é detectado, ou seja, é possível isolá-lo e caracterizá-lo, as reações são consideradas associativas ou dissociativas. Quando não é possível isolar o intermediário, as reações são de intercâmbio, podendo ter caráter associativo (Ia) ou dissociativo (Id). A seguir serão dados maiores detalhes com relação aos diferentes mecanismos.[7]
Associativo (A): a ligação M-X é totalmente formada antes que a ligação M-L comece a ser quebrada. É um mecanismo bastante comum para complexos com baixos números de coordenação, por exemplo, NC=4. Assim, o intermediário possui número de coordenação uma unidade maior que o complexo inicial. Exemplo:
ML4 + X → ML4X → ML3X + L
[NC=4 → NC=5 → NC=4]
Intercâmbio associativo (Ia): a ligação M-L começa a se quebrar antes que a ligação M-X seja totalmente formada, porém a formação de M-X é mais importante que a quebra de M-L. Exemplo:
ML4 + X → X-ML3••L → ML3X + L
Dissociativo (D): a ligação M-L é totalmente quebrada antes do início da formação da ligação M-X. É mais comum em complexos com altos números de coordenação, por exemplo, NC=6. Assim, o intermediário possui número de coordenação uma unidade menor que o complexo inicial. Exemplo:
ML6 + X → ML5 + L + X → ML5X + L
[NC=6 → NC=5 → NC=6]
Intercâmbio dissociativo (Id): a ligação M-X começa a se formar antes da quebra completa da ligação M-X, porém a quebra da ligação M-L é mais importante que a formação da ligação M-X. Exemplo:
ML6 + X → ML5•••L + X → ML5X + L
Efeito trans |
O termo efeito trans (ver isomeria em complexos de número de coordenação quatro) foi utilizado pela primeira vez para compostos de platina(II).[5] Por exemplo, considerando a cisplatina, um complexo de geometria quadrada (ou quadrado planar), ligantes trans aos cloretos são mais facilmente substituídos do que os ligantes trans às amônias. Como exemplo tem-se a reação de [PtCl4]2- com NH3 e a reação de [Pt(NH3)4]2+ com Cl-, como mostrado na figura a seguir.
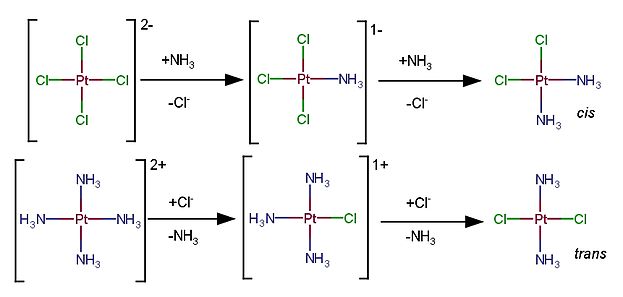
No caso do [PtCl4]2-, com a adição da primeira amônia a segunda irá substituir um cloreto em que há outro cloreto em posição trans. Já no caso do [Pt(NH3)4]2+ a adição do primeiro cloreto faz com que a amônia em posição trans a ele seja labilizada. Diz-se que o cloreto é um ligante direcionador trans mais forte que a amônia. O efeito trans é um efeito cinético, ou seja, ele influencia a velocidade de substituição devido à labilização dos ligantes em posição trans a um ligante direcionador. Direcionadores mais fortes tendem a fazer com que as reações de substituição sejam mais rápidas.[7]
Dois fatores importantes influenciam o efeito trans: o enfraquecimento da ligação M-L (L sendo o ligante de saída) e a estabilização do intermediário no estado de transição.
A labilização do ligante L depende da força da ligação M-L, que é influenciada pelo ligante trans (ligante T). Os dois ligantes, L e T, compartilham os mesmos orbitais do metal, os orbitais px e dx2-y2 (ligação T-M-L). Quando a ligação T-M é muito forte, a ligação M-L é mais fraca (como a brincadeira do cabo de guerra, o ligante T “puxa mais” o metal que o ligante L). Ou seja, a ligação σ M-L é maior em energia quando existe um forte ligante T (estado fundamental é maior em energia), o que leva a uma menor energia de ativação para a quebra dessa ligação M-L. Esse efeito no estado fundamental da ligação σ é um efeito termodinâmico, denominado de influência trans. Ele altera o comprimento da ligação M-L, interferindo na dissociação do ligante de saída. Apesar de ser um efeito termodinâmico, ele afeta a cinética como um todo. Muitos efeitos cinéticos podem estar relacionados a efeitos termodinâmicos, por exemplo, a força de uma ligação metal-ligante (função termodinâmica) pode possuir papel importante na velocidade de dissociação desse ligante (função cinética).[5] É exatamente o caso da influência trans no efeito trans.
Com relação ao caráter σ-doador dos ligantes, eles seguem a seguinte ordem:
H- > PR3 > SCN- > I- ~ CO ~ CN- > Br- > Cl- > NH3 > OH-
É interessante observar que essa série não representa o efeito trans como um todo, pois os ligantes CO e CN-, que se pode dizer que são os ligantes que exercem maior efeito trans, estão no meio da série. Como já comentado, essa série representa apenas o caráter σ-doador dos ligantes, que é apenas um dos fatores que influenciam o efeito trans.[5]
O segundo fator que influencia o efeito trans é a estabilização do intermediário no estado de transição. Em complexos quadrados, o mecanismo de substituição é, na maioria das vezes, associativo, e se dá pela formação de um intermediário no estado de transição de número de coordenação 5, de geometria bipirâmide trigonal, como mostrado na figura a seguir.
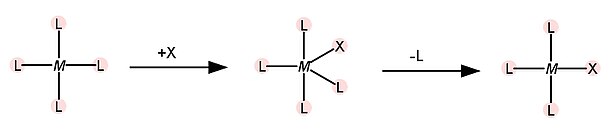
Quando o ligante T é um forte π aceptor, há retirada de densidade eletrônica do metal, o que facilita a coordenação do ligante de entrada. Os orbitais dx2-y2, dxz e dyz do metal podem contribuir para a ligação π no intermediário bipirâmide trigonal. Nesse caso, não há influência na energia do ligante de entrada e sim na energia do intermediário no estado de transição. Ocorre uma diminuição na energia do intermediário, o que diminui a energia de ativação para a reação de substituição, afetando a cinética. A ordem de caráter π-aceptor dos ligantes é a seguinte:
CN- ~ CO ~ C2H4 > NO2- > SCN- > I- > Br- > Cl- > NH3 > OH-
Assim, a ordem expandida da força de direcionamento trans dos ligantes é a seguinte:
CN- ~ CO ~ C2H4 > PR3 ~ H- > NO2- ~ I- ~ SCN- > Br- > Cl- > NH3 > OH- > H2O
Os ligantes do início da série são fortes π-aceptores, seguidos de fortes σ-doadores (exercem influência trans).[5]
Estabilidade cinética e termodinâmica |
É importante diferenciar os termos estável e instável dos termos lábil e inerte em química de coordenação. Se a formação de um composto é termodinamicamente favorável, dizemos que ele é estável. Se não é favorável, ele é instável. Assim, os termos estável e instável se referem à termodinâmica. Agora, se a substituição de ligantes em um composto é rápida, dizemos que o composto é lábil. Se a substituição não é rápida, dizemos que ele é inerte. Dessa forma, os termos lábil e inerte são relacionados à cinética.
Substituição em compostos octaédricos |
Os íons metálicos foram separados em classes com relação à velocidade de substituição de H2O, como mostrado na tabela a seguir.

Os compostos da primeira classe são regidos essencialmente por forças eletrostáticas, incluindo os metais alcalinos e os alcalinos terrosos de maior raio. Esses íons metálicos são caracterizados por possuírem baixa carga e alto raio. Os compostos da segunda classe são metais de transição com carga 2+ e lantanídeos com carga 3+. Nesses complexos a energia de estabilização de campo ligante é baixa. Os compostos da terceira classe são em maioria metais de transição com carga 3+, com energia de estabilização de campo ligante intermediária. Por fim, os compostos da quarta classe são inertes e possuem alta energia de estabilização de campo ligante.[7]
Assim, para o estudo de reações de substituição em compostos octaédricos os íons metálicos mais utilizados são Cr(III) (d3) e Co(III) de baixo spin (d6), por serem inertes e permitirem o acompanhamento dos intermediários das reações.
A reação geral para compostos octaédricos [ML6 + X → produtos] pode ser associativa (A ou Ia) ou dissociativa (D ou Id). No geral, mecanismos dissociativos são mais favoráveis, pois em mecanismos associativos o estado de transição passaria por um intermediário de NC=7, o que é bastante dificultado por efeitos estéreos. Dessa forma, dois casos são observados para a reação geral: em altas concentrações de X, a velocidade de substituição é independente de X, apontando para um mecanismo dissociativo, ou em baixas concentrações de X a velocidade de substituição depende de X e ML6, sugerindo um mecanismo associativo. Assim, essa aparente contradição é explicada pelo mecanismo Eigen-Wilkins, que afirma que há primeiramente a formação de um “complexo de encontro” {ML6,X} entre o complexo octaédrico (ML6) e o ligante de entrada (X) em uma etapa de pré-equilíbrio, com a seguinte formação dos produtos na etapa lenta da reação.[4] Esse mecanismo é uma hipótese para determinação de uma constante de pré-equilíbrio. Essa constante de pré-equilíbrio, juntamente com a constante de velocidade observável da reação permite a formulação de um modelo que pode predizer o tipo de mecanismo de substituição.[8]
Outros mecanismos de reação em compostos de coordenação – reações redox |
As reações de substituição em compostos de coordenação ocorrem sem a mudança no estado de oxidação do metal. Caso haja mudança no estado de oxidação, as reações são de oxi-redução ou redox. Neste caso, são reações de transferência de elétrons, em que os mecanismos podem ser de esfera interna ou de esfera externa.
Técnicas de caracterização de compostos de coordenação |
Quando um composto de coordenação ou complexo metálico é sintetizado, sua composição química, estrutura química e propriedades podem ser investigadas utilizando-se as técnicas de caracterização a seguir:
Análise elementar (CHN) ou microanálise |
A análise elementar ou Microanálise é um método rápido e de baixo custo para a determinação da pureza de um composto de coordenação. O princípio da técnica é baseado na combustão de uma amostra em altas temperaturas (~900°C) na presença de oxigênio. Através da detecção de gases resultantes da decomposição do composto, o percentual em massa dos elementos C, H e N são obtidos. O preparo de amostra exige que o composto seja seco, estando livre de solventes, até massa constante para fornecer resultados confiáveis.
Uma aplicação da análise elementar em química de coordenação seria, por exemplo a determinação da pureza e do teor de água do complexo Na3[Fe(CN)5NH3].xH2O O complexo apresenta as seguintes porcentagens experimentais C:18,5%, H: 2,6% e N: 26,0%. Obtendo-se a fórmula percentual do complexo, as porcentagens teóricas de CHN seriam iguais a 22,01%, 1,11% e 30,90% (Tabela 1). Entretanto, considerando-se x=3 moléculas de H2O, os percentuais de carbono, hidrogênio e nitrogênio são iguais a 18,42%, 2,78% e 25,78%, respectivamente, como na Tabela 2. A proximidade entre os valores experimentais e teóricos é um indicativo da pureza do complexo.

Difratometria ou difração de raios X de pó (DRX) |
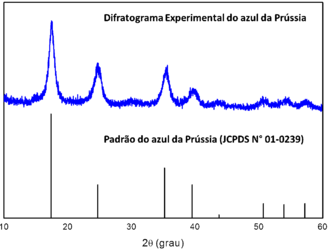
A difração de raios X de pó pode ser utilizada para a identificação de amostras desconhecidos devido à existência de um banco de dados (Joint Committee on Powder Diffraction Standards – JCPDS) de todos os compostos já caracterizados por essa técnica. A fase cristalina, o tamanho do cristalito, as distâncias entre os planos e até mesmo a estrutura cristalina podem ser obtidos por DRX. O banco de dados também pode ser utilizado para confirmar a pureza de um composto de coordenação, como mostrado na Figura 1, em o difratograma do azul da Prússia é consistente ao padrão JCPDS (N° 01-0239).
Difratometria de raios X em monocristal |
A difratometria de raios X em monocristais é uma das técnicas mais importantes para a obtenção da estrutura de compostos de coordenação. O sistema cristalino, as posições de todos os átomos que constituem o complexo, os comprimentos e os ângulos de ligação podem ser determinados. As interações intermoleculares e as posições relativas de todas as moléculas que compõem a cela unitária também podem ser obtidas.
Espectroscopia eletrônica na região do UV/visível |
A espectroscopia UV/visível está relacionada à transição eletrônica molecular que ocorre quando a energia absorvida por uma molécula é utilizada para excitar um elétron para um nível de maior energia. Essa técnica é bastante estudada para compostos coloridos como grande parte dos compostos de coordenação, que absorvem nessa região do espectro.
Um espectro UV-Vis ou espectro eletrônico pode apresentar transições d-d, ou seja, que ocorrem entre os orbitas d do metal; transições de transferência de carga do metal para o ligante e do ligante para o metal; e transições intraligantes e transições de intervalência (que ocorrem em compostos com dois centros metálicos redox, como o azul da Prússia). Os valores de absorbância das bandas em um espectro eletrônico estão relacionados à concentração da amostra, ao caminho óptico e ao coeficiente de absorbtividade molar (Lei de Beer-Lambert).
De acordo com a teoria do campo cristalino, a substituição de um determinado ligante em um complexo pode causar alterações na sua cor e o deslocamento de uma transição d-d em um espectro eletrônico, dependendo do valor do desdobramento do campo cristalino (Δo) da força do campo que varia de acordo com a série espectroquímica como mostrado na Figura 2.[9]

Além das informações sobre a estrutura eletrônica dos compostos de coordenação, estudos de cinética química em complexos e a determinação de parâmetros termodinâmicos de uma dada reação podem ser obtidos. Por exemplo, a cinética de substituição de ligantes N-heterocíclicos em complexos pentacianoferratos por dimetilsulfóxido (DMSO) pode fornecer informações sobre o mecanismo de substituição que pode ser dissociativo, associativo, intercâmbio dissociativo ou intercâmbio associativo. No caso da cinética de substituição de um ligante N-heterocíclico (L) por DMSO a velocidade independe da concentração do DMSO, mas depende da saída do ligante L, característico de um mecanismo dissociativo. Os parâmetros termodinâmicos podem ser obtidos realizando-se a cinética de substituição em diferentes temperaturas.[10]
A partir das curvas cinéticas, constantes de dissociação em diferentes temperaturas podem ser obtidos e através da equação de Eyring apresentada abaixo, valores de entalpia e entropia de dissociação podem ser determinados.
lnkT=−ΔH‡R⋅1T+lnkBh+ΔS‡R{displaystyle ln {frac {k}{T}}={frac {-Delta H^{ddagger }}{R}}cdot {frac {1}{T}}+ln {frac {k_{mathrm {B} }}{h}}+{frac {Delta S^{ddagger }}{R}}}

Onde: k é a constante de velocidade da reação, T é a temperatura, ΔH≠ é a entalpia de ativação da reação de dissociação, R é a constante dos gases, kB é a constante de Boltzmann, h é a constante de Planc e ΔS≠ é a entropia de dissociação.
Realizando-se o estudo de cinético de uma reação, o número de espécies pode ser determinado e pelo ponto isosbético é possível avaliar se duas espécies estão em equilíbrio.
Espectroscopia Vibracional na Região do Infravermelho e Espectroscopia Raman |
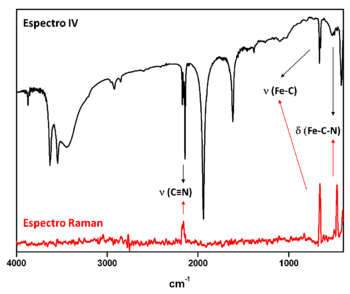
As técnicas de espectroscopia vibracional podem ser utilizadas para a identificação de um composto de coordenação em uma amostra desconhecida, para a obtenção de informações estruturais e a determinação de grupos funcionais presentes nesses compostos, para o estudo cinético de reações químicas e para a identificação de produtos de reação. As técnicas de espectroscopia de infravermelho (IV) e espectroscopia Raman podem ser consideradas complementares, sendo que alguns modos vibracionais podem ser ativos em uma técnica e inativos na outra ou podem ser ativos em ambas. Os modos vibracionais que levam à mudança no momento de dipolo são ativos no IV e apenas as vibrações que causam mudança na polarizabilidade são ativas no Raman.
Por exemplo, nos espectros vibracionais de um complexo [Fe(CN)5L]n- (Figura 3), os modos vibracionais que ocorrem próximos a 2100 cm-1(ν C≡N), 660 cm-1 (ν Fe-C), e 450 cm-1 (δ Fe-C-N) são ativos em ambas as técnicas, enquanto que os demais modos são inativos no Raman.
Voltametria ou voltamperometria cíclica |
A voltametria ou voltamperometria cíclica é uma técnica que permite o estudo das propriedades eletroquímicas dos compostos de coordenação. Os processos redox característicos de um complexo e os potenciais em que ocorrem podem ser determinados. Informações sobre a pureza do produto de uma síntese, os produtos de reações, a reversibilidade de um processo redox, a cinética de transferência eletrônica e sobre o coeficiente de difusão do composto de interesse também podem ser obtidas.
Por exemplo, o potencial de oxidação (E1/2) do complexo [Fe(CN)6]4- pode ser calculado pela média dos valores do potencial de pico anódico (Epa) e do potencial de pico catódico (Epc) mostrados na Figura 4.
![Figura 4: Voltamogramas de uma solução aquosa do complexo [Fe(CN)6]4-.](http://upload.wikimedia.org/wikipedia/commons/thumb/b/b4/Sup-..png/450px-Sup-..png)
O coeficiente de difusão (Do) pode ser determinado pela equação de Randles–Sevcik, quando a área real do eletrodo é conhecida.
ip=268600 n32AD12Cv12{displaystyle i_{p}=268600 n^{frac {3}{2}}AD^{frac {1}{2}}Cv^{frac {1}{2}}}

Onde: ip é a corrente do pico anódico (A), n é o número de elétrons envolvidos no processo, ν a velocidade de varredura (V/s), Co* é a concentração do material oxidado (mol/cm3) e Do é o coeficiente de difusão do composto de coordenação (cm/s).
A voltametria cíclica também pode ser utilizada para estudar o efeito doador ou receptor dos ligantes, sendo que o potencial de oxidação de um complexo, um pentacianidoferrato( [Fe(CN)5L]n-), por exemplo, aumenta em função da característica π-receptora do ligante, o que dificulta a oxidação do ferro. Comparando-se dois complexos [Fe(CN)5L’]n- e [Fe(CN)5L”]m- em que o ligante L” tem maior caráter π-receptor, o que apresenta o maior potencial de oxidação é o [Fe(CN)5L”]m-(Figura 5).

Referências
↑ SIRIWARDANE, Upali; HALL, Carson Taylor. CHEM 102 CLASS NOTES: Chapter 7 Arquivado em 27 de setembro de 2007, no Wayback Machine.. d-Metal Complexes. Acesso em: 5 out. 2008.
↑ Kauffman, George B.; Halpern, Jack (15 de agosto de 2008). «Coordination compound | chemistry». Encyclopedia Britannica (em inglês). Consultado em 11 de junho de 2018
↑ abcdefg JONES, Chris J. A Química dos Elementos dos Blocos d e f, Porto Alegre, Editora Bookman, 2002 ISBN 8-573-07977-0 OCLC 55906222
↑ ab C.E. Housecroft, A.G. Sharpe, Inorganic Chemistry, 2nd edition, Pearson Education Limited.
↑ abcde G.L. Miessler, D.A. Tarr, Inorganic Chemistry, 3rd edition, Pearson Education International.
↑ H. Taube, Rates and Mechanisms of Substitution in Inorganic Complexes in Solution, Chemical Reviews 50 (1951) 69-126.
↑ abc J.E. Huheey, E.A. Keiter, R.L. Keiter, Inorganic Chemistry: Principles of Structure and Reactivity, 4th edition, Harper Collins College Publishers.
↑ D. Shriver, P. Atkins, Inorganic Chemistry, 5th edition, Oxford University Press.
↑ GUSHIKEM, YOSHITAKA. Espectros eletrônicos de alguns complexos de geometria octaédrica de Ni2+: Uma introdução prática à teoria do campo cristalino no curso de graduação. Quim. Nova, v. 28, p. 153-156, 2005
↑ MORANDI PIRES, BRUNO et al. Prussian Blue Films Produced by Pentacyanidoferrate (II) and Their Application as Active Electrochemical Layers. European Journal of Inorganic Chemistry, v. 2014, n. 34, p. 5812-N5819, 2014.
Aplicações de compostos de coordenação |
Embora a química de coordenação apresente uma série de desafios, tanto teórico-conceituais quanto práticos, ao estudante que deseje compreender o assunto, além de ter tido um desenvolvimento conceitual relativamente recente (desde o fim do século XIX), é inegável que compostos de coordenação estão envolvidos diretamente com o nosso dia a dia, sendo encontrados não apenas em processos industriais e fármacos, mas também naturalmente nos próprios organismos. Dentre as principais aplicações dos compostos de coordenação pode-se destacar:
Ocorrência natural |
Os metais ocorrem amplamente na natureza na forma de minerais e rochas, como óxidos, silicatos, carbonatos etc., mas podem ser também encontrados, quase sempre em solução, como compostos de coordenação. Os exemplos mais importantes estão envolvidos em sistemas biológicos e fazem parte, portanto, do nosso cotidiano, embora muitas vezes não se tome conhecimento disso.
Transporte de oxigênio |
Uma função fundamental em muitos organismos é o transporte de oxigênio molecular (O2), uma função exclusiva de metaloproteínas. Na espécie humana, assim como em todos os cordados, essa função é realizada pela Hemoglobina e pela Mioglobina, proteínas cujo sítio de ligação do oxigênio contém uma porfirina de ferro. Quando o O2 se liga a estas proteínas, importantes mudanças estruturais ocorrem, sem, contudo, levar a reações de oxirredução irreversíveis, permitindo que o oxigênio seja liberado nas condições adequadas. Outra metaloproteína transportadora de oxigênio é a hemocianina, encontrada em artrópodes e moluscos. Nela, dois átomos de cobre são coordenados por três resíduos de histidina e atuam em conjunto.[1]
Fotossíntese |
Vários organismos autótrofos captam energia luminosa através da clorofila, um pigmento verde que contém uma porfirina de magnésio. Esse pigmento é capaz de absorver luz, passando a um estado excitado em que um elétron é transferido em cascata por uma série de moléculas, incluindo algumas metaloproteínas, até o NADP. Por outro lado a clorofila recebe elétron de volta de uma proteína contendo um cluster de manganês que, por sua vez, reduz água a gás oxigênio.[2]
Vitamina B12 |
A Vitamina B12 foi isolada de extrato de fígado pela primeira vez em 1948. Foi o primeiro exemplo de composto organometálico de ocorrência natural. Ela participa na atuação de enzimas, liases e mutases, que catalisam reações de deslocamento 1,2. Esse tipo de reação dificilmente acontece sem a participação de um catalisador.[2]
Extração de metais menos comuns |
Alguns metais menos comuns são apenas encontrados em quantidades minoritárias, misturados a outros minérios. Sua obtenção pode se dar, entre outros métodos, por hidrometalurgia, na qual um complexo do metal de interesse é formado e se dissolve, enquanto outros metais presentes no minério (ou muitas vezes em lamas anódicas) não são dissolvidos. Um exemplo desse processo é a hidrometalurgia do ouro pelo processo do cianeto. Tradicionalmente, o ouro foi recolhido de areias fluviais pelo uso de bateias, levando em conta sua elevada densidade. Contudo, como estas fontes já foram extensamente exploradas a produção atual depende da mineração de rochas com tipicamente entre 5 e 15 ppm de ouro. Para a extração os minérios são triturados até um pó fino, que é extraído com uma solução aerada de cianeto de sódio.[3]
4Au + 8NaCN + O2 + 2H2 O → 4Na[Au(CN)2] + 4NaOH[3]
Outros exemplos comuns são a clorinação de Newbery-Vautin para extração de ouro e o Processo Bayer de extração de Alumínio a partir da Bauxita.
Pigmentos e corantes |
A maior parte dos pigmentos e corantes inorgânicos são materiais, tais como o Vermilion (sulfeto de mercúrio (II)), o Amarelo de Cromo (Cromato de Chumbo (II)), o Verde de Cromo (óxido de cromo (III)), a Malaquita (carbonato básico de cobre (II)) os Ocres (diversos óxidos de ferro), o Azul Egípcio (silicato de cobre(II) e cálcio), etc. Um caso particular interessante é o Azul da Prússia, um material obtido partir do complexo hexacianoferrato(II), precipitado com ferro(III).[3] Contudo, algumas classes de compostos de coordenação são amplamente utilizadas como corantes, tais como as ftalocianinas azuis de Fe(II), Co(II), Ni(II), Cobre(II) e Zn(II) e azocomplexos de ferro e cobre.
Catálise |
A aplicação de catalisadores baseados em metais de transição é absolutamente fundamental na produção de grande parte das substâncias químicas industrialmente relevantes. Reações catalisadas têm a dupla vantagem de permitir sínteses mais ágeis e com menores custos. São importantes inclusive do ponto de vista da sustentabilidade, já que, em geral, permitem condições mais brandas de reação, dependendo de menor gasto de energia e produzindo menor quantidade de resíduos ou, pelo menos, resíduos mais brandos.
A maior parte dos catalisadores utilizados são metais no estado elementar ou óxidos metálicos em processos heterogêneos. Contudo a partir dos anos 1960 catalisadores baseados em compostos de coordenação, principalmente organometálicos, sofreram uma grande expansão conduzida principalmente por três processos industrialmente importantes: Ziegler, Wacker e Oxo. Esses são processos de catálise homogênea.[2]
Ziegler |
Catalisadores de Ziegler são usados na polimerização de olefinas. Eles costumam ser complexos derivados de metais dos grupos 4, 5 e 6 como titânio, vanádio, cromo e zircônio em combinação com promotores organometálicos, por exemplo, trietilalumínio.[2]
Wacker |
Compreende a oxidação parcial de uma olefina em presença de [PdCl4]2- e um cooxidante, geralmente sais de cobre(I). É fundamental na produção de acetaldeído a partir do etileno.[2]
Oxo |
Um dos mais importantes processos industriais envolvendo catálise por complexos de metais de transição, o processo Oxo permite a adição de hidrogênio e monóxido de carbono à olefinas (hidroformilação). Nesse processo são utilizados catalizadores baseados em ródio com ligantes fosfinas ou cobalto com ligantes CO além de, em ambos os casos, um ligante hidreto.[2]
Ativação de pequenas moléculas |
Um esforço bastante grande tem sido feito na comunidade científica no sentido de encontrar rotas catalíticas que favoreçam reações chamadas, genericamente, de ativação de pequenas moléculas. Moléculas pequenas muito pouco reativas como água, gás nitrogênio e dióxido de carbono são muito abundantes e podem ser convertidas a espécies de maior utilidade e valor agregado como gás hidrogênio, amônia e formol, respectivamente, dentre diversos outros produtos através de reações de custo energético muito elevado. Existem diversas propostas de catalisadores homogêneos que envolvem compostos de coordenação que possam permitir condições mais brandas, baratas e ambientalmente amigáveis. Como exemplo, pode-se citar os complexos triamidomolibdênio(III) e triamidonióbio(III) que permitem a conversão de nitrogênio em amônia (utilizada em fertilizantes e explosivos), ou os diversos complexos mono e bimetálicos de rutênio-oxo que atuam na quebra da água a O2 e H2 (utilizado como combustível limpo).
Química medicinal |
O uso de íons metálicos está presente em práticas médicas desde a antiguidade.[4] Os registros mais antigos mostram que os chineses já utilizavam ouro em 2500 a.C,[5] mas outras civilizações do mundo antigo como gregos, egípcios e hindus também utilizaram este metal, assim como a prata, em suas preparações para curas. Essas preparações, contudo, foram elaboradas numa perspectiva muito mais mágico/mística do que científica.
Os compostos metálicos utilizados hoje na clínica podem ser divididos em dois grupos principais por suas aplicações: terapêuticos ou diagnósticos. No que diz respeito a agentes diagnósticos, podem ser citados como exemplos o sulfato de bário, que é usado há pelo menos 100 anos como contraste em radiografia, compostos de gadolínio que são utilizados como contraste para ressonância magnética nuclear e complexos de 99mTc, 67Ga e 111In desenvolvidos e aprimorados para técnicas de rádio imagem (como as diversas cintilografias).[4]
O primeiro caso de um estudo sistemático de composto de ouro dando origem a uma substância terapêutica data de 1890. Koch[5] observou uma inibição no crescimento de bacilos causadores de tuberculose causada pelo dicianoaurato(I), dando origem a investigação de compostos de ouro(I) úteis na clínica. Nos anos seguintes o uso esporádico desses compostos no tratamento da tuberculose levou a observação de que seus efeitos se estendiam a artrite reumatóide. Os tiolatos de ouro compreendem os primeiros compostos dessa longa série de medicamentos para artrite baseados em ouro, culminando em 1976 na auranofina,[6] um fármaco administrado por via oral ainda disponível no mercado. Não apenas sobre artrite, foram reportadas também evidências de atividade da auranofina sobre outras doenças, com destaque para a atividade anti-HIV,[7][8] assim como antibacteriana9, embora os mecanismos de ação dessa droga não sejam conhecidos.
Porém, talvez um dos mais importantes fármacos inorgânicos tenha sido descoberto “casualmente” durante a década de 1960: a cis-diaminodicloroplatina(II), frequentemente denominada cisplatina. Este é um potente quimioterápico para o combate de células cancerígenas, particularmente o câncer de testículo. Sua importância é tão grande que, atualmente, a mortalidade por câncer de testículo é inferior a 5%. Antes de sua introdução, em 1971, a mortalidade ultrapassava 90%.[4] Complexos análogos foram introduzidos na clínica para minimizar os efeitos adversos inerentes à cisplatina, como a carboplatina e a oxaliplatina,[9] após intensos trabalhos ao redor de todo o mundo para a compreensão de seu mecanismo de atuação.
Ver também |
- Nomenclatura IUPAC de compostos inorgânicos
- Geometria de coordenação
- Química organometálica
- Compendium of Chemical Terminology
Bibliografia |
- SHRIVER, DUWARD; ATKINS, PETER. Química inorgânica - 4ª edição. Porto Alegre, Bookman, 2008. ISBN 0-198-50331-8 ISBN 0-198-50330-X OCLC 40473750
- LEE, J.D.. Química inorgânica não tão concisa – tradução da 4ª edição inglesa. São Paulo, Edgard Blücher, 1996. OCLC 816987467
- BARROS, HAROLDO L.C..Química inorgânica; uma introdução. Belo Horizonte: GAM; Editora distribuidora, 2001
- AYALA, J. D. Química de Coordenação 1. Acesso em: 20 set. 2008.
- VON ZELEWSKY, A. Stereochemistry of Coordination Compounds. United Kingdom: John Wiley & Sons, 1995. 3 v. (Inorganic Chemistry: A Textbook Séries). ISBN 0-471-95057-2 ISBN 0-471-95599-X OCLC 32548965 (em inglês)
- KIPROF, Paul. Coordination Number 4. Acesso em: 20 set. 2008.
- HESLOP, R B; ROBINSON, P.l. Inorganic Chemistry: A Guide to Advanced Study. Amsterdam: Elsevier Publishing Company, 1976. 830 p. OCLC 541332 (em inglês)
- Lecturer in Chemistry, The Manchester College of Science and Technology And P. L. ROBINSON.
- MULLER, Ulrich. Inorganic Structural Chemistry. 2 nd Germany: John Wiley & Sons, 1996. (Inorganic Chemistry: A Textbook Séries). ISBN 0-471-93379-1 OCLC 26161852} (em inglês)
- ATKINS, P. W.; SHRIVER, D. F.. Inorganic Chemistry. 3th Oxford: University Press, 1999. 763 p. ISBN 0-199-28859-3 OCLC 874419900 (em inglês)
- Huheey, J.E., Keiter, E.A., Keiter, R.L., Inorganic Chemistry – Principles of Structure and Reactivity, 4ª ed. Harper Collins, 1993. ISBN 0-060-42995-X OCLC 473106662 (em inglês)
- Ian S. Butler, John F. Harrod, Inorganic Chemistry, principles and applications, Redwood City, Calif. : Benjamin/Cummings, 1989. ISBN 0-805-30247-6 OCLC 18907722 (em inglês)
- RÍOS, Enrique Gutiérrez. QUIMICA INORGÁNICA. Editorial Revérte S.a, 1984. ISBN 8-429-17215-7 OCLC 8213105 (em castelhano)
- Cotton, Frank Albert; Geoffrey Wilkinson; Carlos A. Murillo (1999). Advanced Inorganic Chemistry. p. 1355. ISBN 978-0-471-19957-1 OCLC 849256233. (em inglês)
- De Vito, D.; Weber, J. ; Merbach, A. E. “Calculated Volume and Energy Profiles for Water Exchange on t2g 6 Rhodium(III) and Iridium(III) Hexaaquaions: Conclusive Evidence for an Ia Mechanism” Inorganic Chemistry, 2005, Volume 43, pages 858–863. ISSN 0020-1669 (em inglês)
Referências
↑ Lippard, S. J.; Berg, J. M. Principles of Bioinorganic Chemistrz; Universiy Science Books, Ed.; 1994.
↑ abcdef de Farias, R. F. (Org. . Química de Coordenação: fundamentos e atualidades; 2nd ed.; Editora Átomo: Campinas-SP, Brasil, 2009.
↑ abc Greenwood, N. N.; Earnshaw, A. Chemistry of the Elements; 2nd ed.; Elsevier Ltd, 1997.
↑ abc Concepts and Models in Bioinorganic Chemistry; Kraatz, H.-B.; Metzler-Nolte, N., Eds.; Wiley-VCH, 2006.
↑ ab Brown, D. H.; Smith, W. E. ;Chem. Soc. Rev. 1980, 9, 217.
↑ Finkelstein, a E.; Walz, D. T.; Batista, V.; Mizraji, M.; Roisman, F.; Misher, a ;Ann. Rheum. Dis. 1976, 35, 251.
↑ Badley, A. D.; Sainski, A.; Wightman, F.; Lewin, S. R. ;Cell Death Dis. 2013, 4, e718.
↑ Roder, C.; Thomson, M. ;Drugs R. D. 2015, 15, 13.
↑ Culy, C. R.; Clemett, D.; Wiseman, L. R.; C.R., C.; D., C.; L.R., W. ;Drugs 2000, 60, 895.


